14 P
14.1 Talking Glossary: PCR - The Polymerase Chain Reaction (0.5 min)
National Human Genome Research Institute
https://www.genome.gov/genetics-glossary/Polymerase-Chain-Reaction
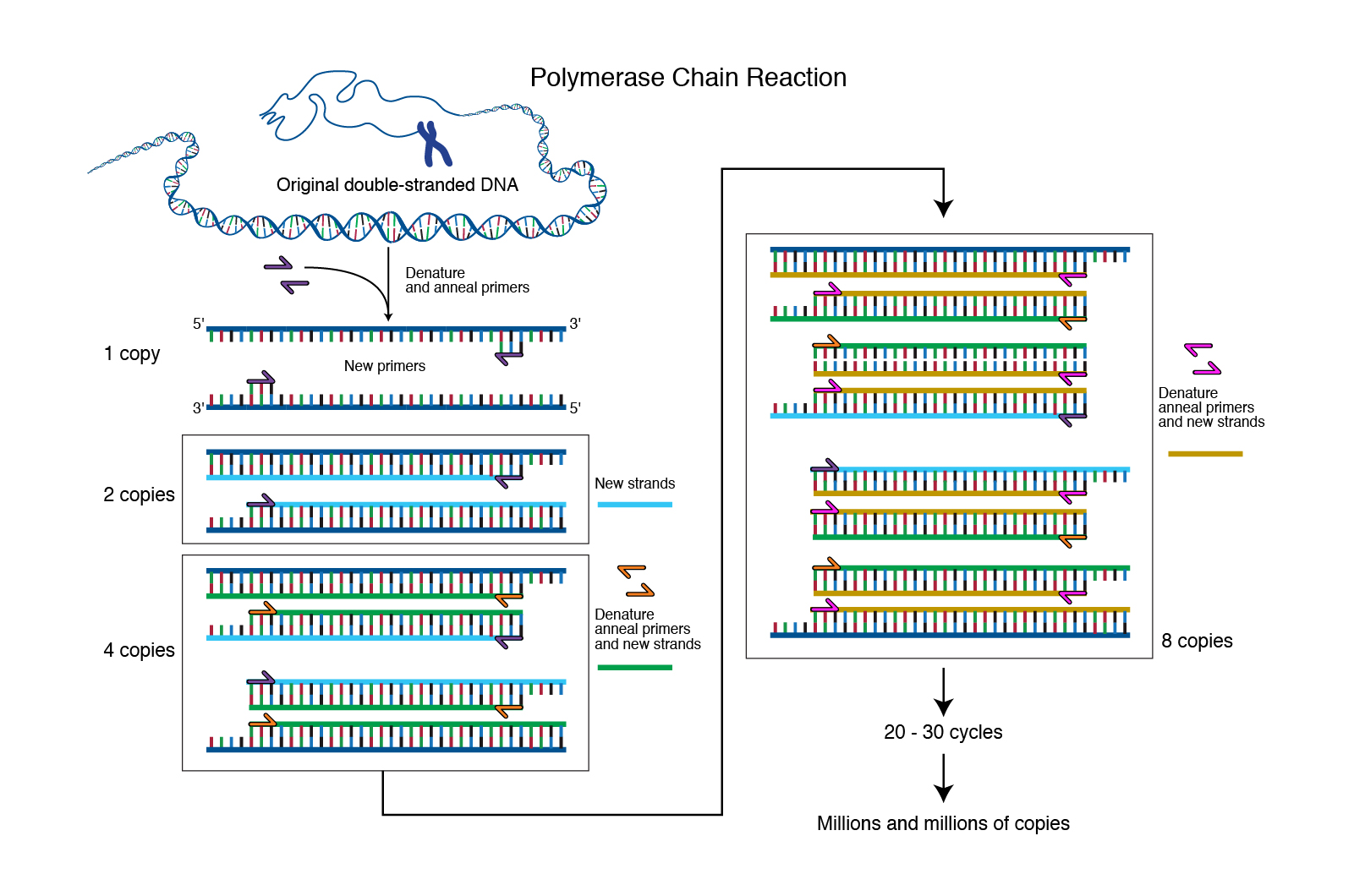
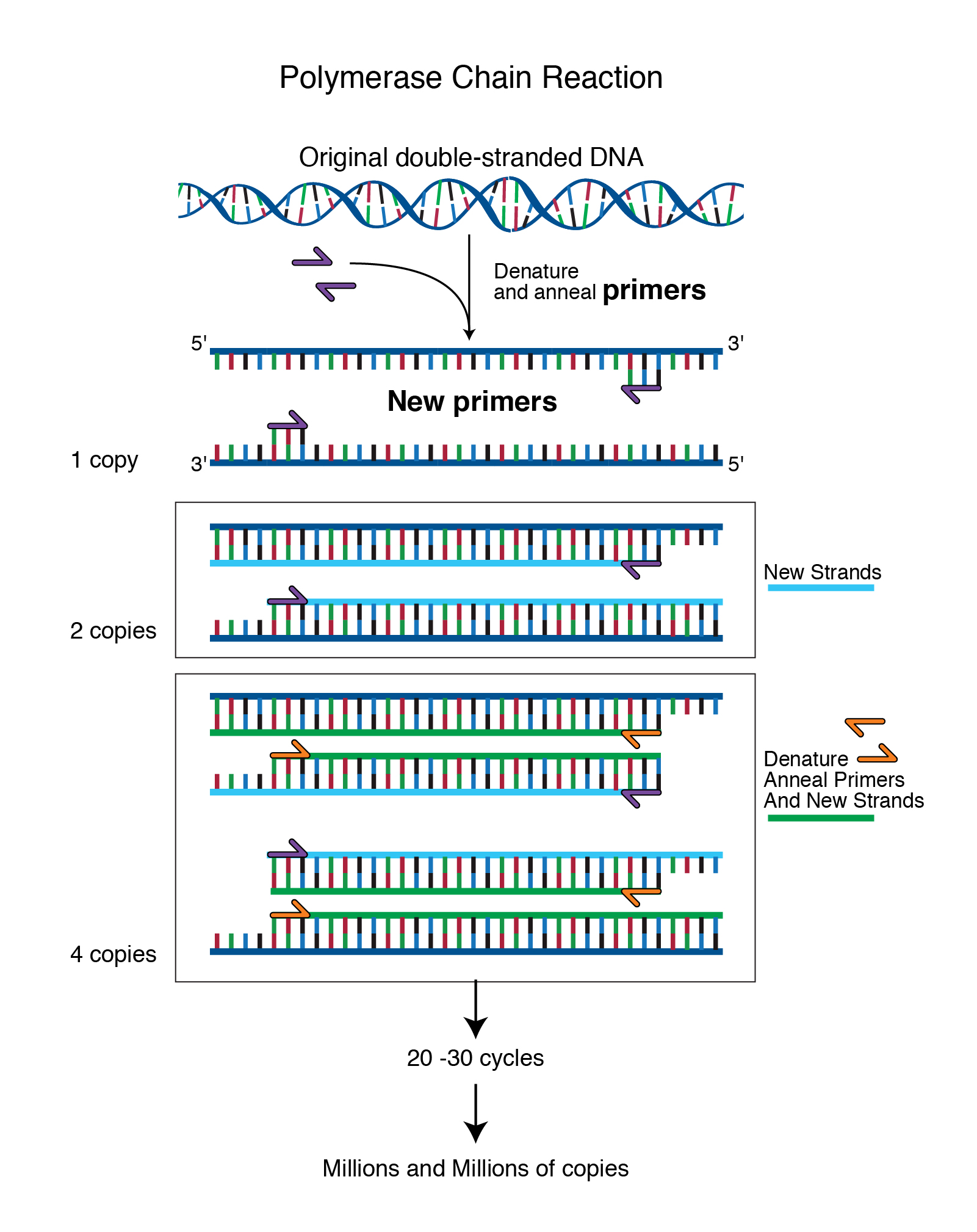
Abstract: Polymerase chain reaction (PCR) is a laboratory technique used to amplify DNA sequences. The method involves using short DNA sequences called primers to select the portion of the genome to be amplified. The temperature of the sample is repeatedly raised and lowered to help a DNA replication enzyme copy the target DNA sequence. The technique can produce a billion copies of the target sequence in just a few hours.
Audio: https://www.genome.gov/sites/default/files/tg/en/narration/polymerase_chain_reaction.mp3
Image: https://www.genome.gov/sites/default/files/media/images/2021-01/polymerase_chain_reaction.jpg
{kind=link}
Transcript: “PCR, or the polymerase chain reaction, is a chemical reaction that molecular biologists use to amplify pieces of DNA. This reaction allows a single or a few copies of DNA to be replicated into millions or billions of copies. And by amplifying that DNA, it allows us to study that DNA molecule in detail in the laboratory.”
Leslie G. Biesecker, M.D.
14.2 Protein Data Bank
Adapated from Wikipedia
The Protein Data Bank (PDB)[1] is a database for the three-dimensional structural data of large biological molecules, such as proteins and nucleic acids. The data, typically obtained by X-ray crystallography, NMR spectroscopy, or, increasingly, cryo-electron microscopy, and submitted by biologists and biochemists from around the world, are freely accessible on the Internet via the websites of its member organisations (PDBe,[2] PDBj,[3] RCSB,[4] and BMRB[5]).
The PDB is a key in areas of structural biology, such as structural genomics. Most major scientific journals and some funding agencies now require scientists to submit their structure data to the PDB. Many other databases use protein structures deposited in the PDB. For example, SCOP and CATH classify protein structures, while PDBsum provides a graphic overview of PDB entries using information from other sources, such as Gene ontology.[6, 7]
Most structures are determined by X-ray diffraction, but about 10% of structures are determined by protein NMR. When using X-ray diffraction, approximations of the coordinates of the atoms of the protein are obtained, whereas using NMR, the distance between pairs of atoms of the protein is estimated. The final conformation of the protein is obtained from NMR by solving a distance geometry problem. After 2013, a growing number of proteins are determined by cryo-electron microscopy. Clicking on the numbers in the linked external table displays examples of structures determined by that method.
Historically, the number of structures in the PDB has grown at an approximately exponential rate, with 100 registered structures in 1982, 1,000 structures in 1993, 10,000 in 1999, and 100,000 in 2014.[19, 20]
14.2.1 History
** OPTIONAL **
Two forces converged to initiate the PDB: a small but growing collection of sets of protein structure data determined by X-ray diffraction; and the newly available (1968) molecular graphics display, the Brookhaven RAster Display (BRAD), to visualize these protein structures in 3-D. In 1969, with the sponsorship of Walter Hamilton at the Brookhaven National Laboratory, Edgar Meyer (Texas A&M University) began to write software to store atomic coordinate files in a common format to make them available for geometric and graphical evaluation. By 1971, one of Meyer’s programs, SEARCH, enabled researchers to remotely access information from the database to study protein structures offline.[8] SEARCH was instrumental in enabling networking, thus marking the functional beginning of the PDB.
The Protein Data Bank was announced in October 1971 in Nature New Biology[9] as a joint venture between Cambridge Crystallographic Data Centre, UK and Brookhaven National Laboratory, US.
Upon Hamilton’s death in 1973, Tom Koeztle took over direction of the PDB for the subsequent 20 years. In January 1994, Joel Sussman of Israel’s Weizmann Institute of Science was appointed head of the PDB. In October 1998,[10] the PDB was transferred to the Research Collaboratory for Structural Bioinformatics (RCSB);[11] the transfer was completed in June 1999. The new director was Helen M. Berman of Rutgers University (one of the managing institutions of the RCSB, the other being the San Diego Supercomputer Center at UC San Diego).[12] In 2003, with the formation of the wwPDB, the PDB became an international organization. The founding members are PDBe (Europe),[2] RCSB (USA), and PDBj (Japan).[3] The BMRB[5] joined in 2006. Each of the four members of wwPDB can act as deposition, data processing and distribution centers for PDB data. The data processing refers to the fact that wwPDB staff review and annotate each submitted entry.[13] The data are then automatically checked for plausibility (the source code[14] for this validation software has been made available to the public at no charge).
For PDB structures determined by X-ray diffraction that have a structure factor file, their electron density map may be viewed. The data of such structures is stored on the “electron density server”.[17, 18]
14.2.2 File format
OPTIONAL The file format initially used by the PDB was called the PDB file format. The original format was restricted by the width of computer punch cards to 80 characters per line. Around 1996, the “macromolecular Crystallographic Information file” format, mmCIF, which is an extension of the CIF format was phased in. mmCIF became the standard format for the PDB archive in 2014.[21] In 2019, the wwPDB announced that depositions for crystallographic methods would only be accepted in mmCIF format.[22]
An XML version of PDB, called PDBML, was described in 2005.[23] The structure files can be downloaded in any of these three formats, though an increasing number of structures do not fit the legacy PDB format. Individual files are easily downloaded into graphics packages from Internet URLs:
14.3 PFam
Adapted from Wikipedia.
Pfam is a database of protein families that includes their annotations and multiple sequence alignments generated using bioinformatics tools known as hidden Markov models (HMM; 1, 2, 3. The most recent version, Pfam 34.0, was released in March 2021 and contains 19,179 families (4).
14.3.1 Uses
The general purpose of the Pfam database is to provide a complete and accurate classification of protein families and domains.[5] Originally, the rationale behind creating the database was to have a semi-automated method of curating information on known protein families to improve the efficiency of annotating genomes.[6] The Pfam classification of protein families has been widely adopted by biologists because of its wide coverage of proteins and sensible naming conventions.[7]
It is used by experimental biologists researching specific proteins, by structural biologists to identify new targets for structure determination, by computational biologists to organise sequences and by evolutionary biologists tracing the origins of proteins.[8] Early genome projects, such as human and fly used Pfam extensively for functional annotation of genomic data.[9, 10, 11]
14.4 Talking Glossary: Phenotype
https://www.genome.gov/genetics-glossary/Phenotype
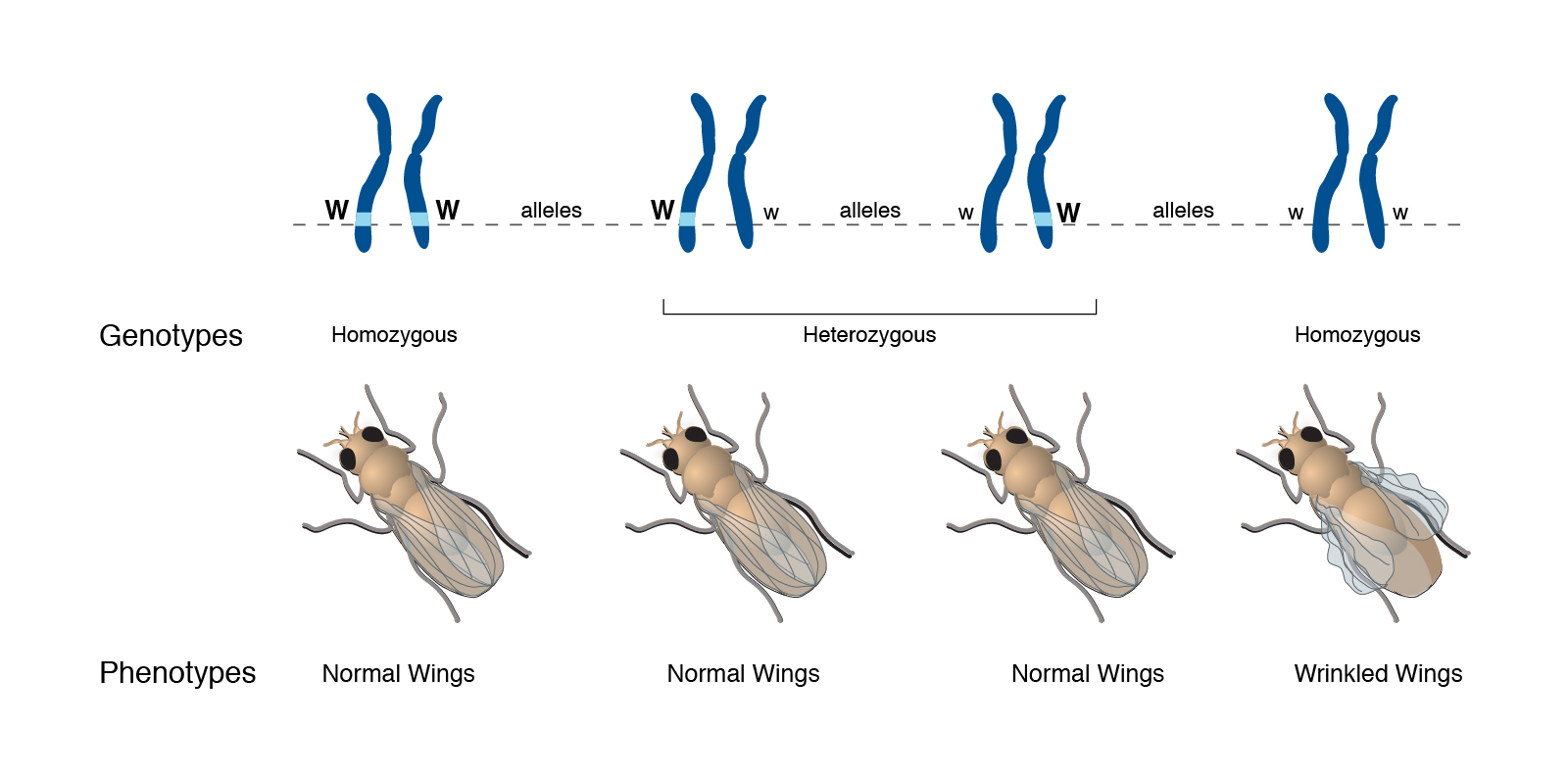
Abstract: “A phenotype is an individual’s observable traits, such as height, eye color, and blood type. The genetic contribution to the phenotype is called the genotype. Some traits are largely determined by the genotype, while other traits are largely determined by environmental factors.”
Audio:
Image: https://www.genome.gov/sites/default/files/tg/en/illustration/phenotype.jpg
{kind=link}
Transcript: ““Phenotype” simply refers to an observable trait. “Pheno” simply means “observe” and comes from the same root as the word “phenomenon”. And so it’s an observable type of an organism, and it can refer to anything from a common trait, such as height or hair color, to presence or absence of a disease. Frequently, phenotypes are related and used–the term is used–to relate a difference in DNA sequence among individuals with a difference in trait, be it height or hair color, or disease, or what have you. But it’s important to remember that phenotypes are equally, or even sometimes more greatly influenced by environmental effects than genetic effects. So a phenotype can be directly related to a genotype, but not necessarily. There’s usually not a one-to-one correlation between a genotype and a phenotype. There are almost always environmental influences, such as what one eats, how much one exercises, how much one smokes, etc. All of those are environmental influences which will affect the phenotype as well.”
Christopher P. Austin, M.D.
14.5 Phylogenetics vocab
Here are definitions of key terms related to phylogenetics.
Apomorphy: A character present in one or more species in a clade that was not present in the clade’s common ancestor; an evolutionary novelty. Also known as a derived character. For a review of ancestral versus derived traits see this video . (Contrast with homplasy).
Autapomorphy: A derived character state that is restricted to one taxon in a particular data set. Apomorphies are often biologically interesting (like wings in birds) but actually have no value for building phylogenetic trees because they don’t let you form clades. Stated another way, autapomorphis are unique to a single taxon and don’t allow you to group taxa into a clade.
Convergence: The multiple and independent appearance in different lineages of similar evolutionary novelties (apomorphies). Often happens due to similar ecological conditions and evolutionary forces.
Convergent evolution: Similarity between species that is caused by a similar, but evolutionary independent, response to a common environmental problem (Figure 1). Convergence can also occur at the molecular level where the same mutation occurs independently in two different lineages (Figure 2).
Derived character: A character present in one or more species in a clade that was not present in the clade’s common ancestor; an evolutionary novelty; also known as an apomorphy. (Contrast with ancestral character)
Homologous: Describes characters derived from a common ancestor. (Contrast with analogous).
Homology: Similarity between species that results from inheritance of traits from a common ancestor. (Contrast with analogy). For more on homologies see Understanding Evolution: Homologies, Homologies: anatomy , Homologies: comparative anatomy, and Homologies: development .
Homoplasy: Technical term which indicates similarity in the characters/traits found in different species that are unrelated to common ancestry. Instead, they can be due to convergent evolution or reversal - not common descent. An analogous trait is a type of homoplasy. For an in-depth discussion of homology versus homoplasy see this video (Contrast with homology).
Lineage: A group of ancestral and descendant populations, species, or other taxa that are descended from a common ancestor. Synonymous with clade.
Outgroup: A taxonomic group that diverged prior to the rest of the focal taxa (focal clade) in a phylogenetic analysis.
Parsimony: A criterion for selecting among alternative patterns or explanations based on minimizing the total amount of change or complexity. Can be approximate as “simpler answers are to be preferred.”
Reversal / evolutionary reversal: An event that results in the reversion of a derived trait (apomorphy) to the ancestral form. This happens frequently at the DNA level, where a mutation can change a base to something different, then later another mutation changes it back. This can be called a back mutation. Reversals can also occur at the protein or morphological level.
Trait matrix (aka character matrix, character state matrix, data matrix): A table showing the state of each trait occurring in each taxon. Generally, row represents taxa, columns represent characters, and numbers (usually just 0 or 1) represent character states.
14.6 Talking Glossary: Plasmid (1 min)
National Human Genome Research Institute https://www.genome.gov/genetics-glossary/Plasmid
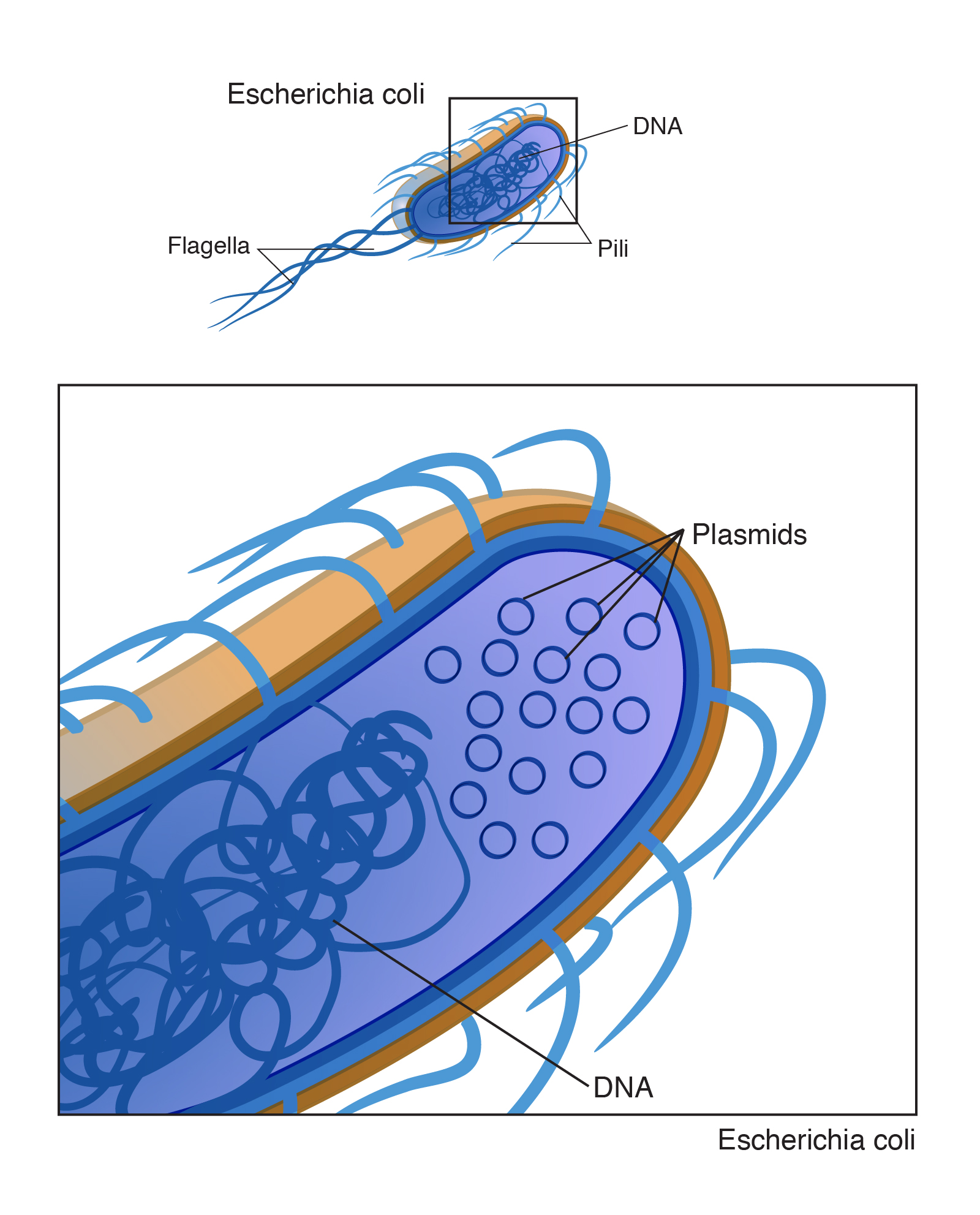
Intro: “A plasmid is a small, often circular DNA molecule found in bacteria and other cells. Plasmids are separate from the bacterial chromosome and replicate independently of it. They generally carry only a small number of genes, notably some associated with antibiotic resistance. Plasmids may be passed between different bacterial cells.”
Image: https://www.genome.gov/sites/default/files/tg/en/illustration/plasmid.jpg
{kind=link}
Audio: https://www.genome.gov/sites/default/files/tg/en/narration/plasmid.mp3
Transcription: “Electrophoresis is a laboratory technique used to separate DNA, RNA, or protein molecules based on their size and electrical charge. An electric current is used to move molecules to be separated through a gel. Pores in the gel work like a sieve, allowing smaller molecules to move faster than larger molecules. The conditions used during electrophoresis can be adjusted to separate molecules in a desired size range.”
14.7 Talking Glossary: Point mutation (0.5 min)
Point mutation
https://www.genome.gov/genetics-glossary/Point-Mutation
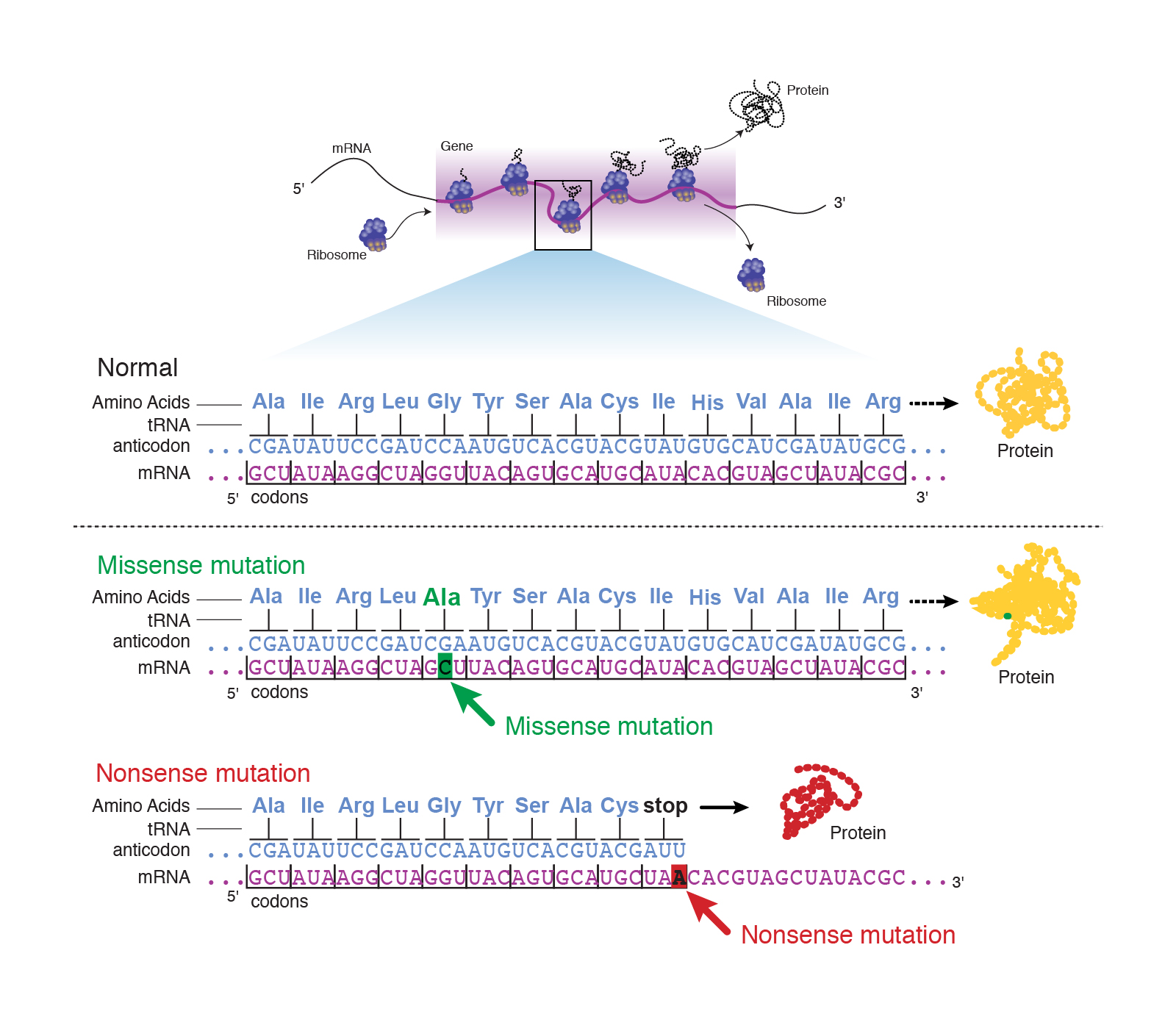
Abstract: “A point mutation is when a single base pair is altered. Point mutations can have one of three effects. First, the base substitution can be a silent mutation where the altered codon corresponds to the same amino acid. Second, the base substitution can be a missense mutation where the altered codon corresponds to a different amino acid. Or third, the base substitution can be a nonsense mutation where the altered codon corresponds to a stop signal.”
Image: https://www.genome.gov/sites/default/files/tg/en/illustration/point_mutation.jpg
{kind=link}
Audio: https://www.genome.gov/sites/default/files/tg/en/narration/point_mutation.mp3
NOTE: Sources vary a little bit in how they define point mutations. Like this glossary entry, I include indels (insertions / deletions) as a type of point mutation.
Transcript: “Point mutations are a large category of mutations that describe a change in single nucleotide of DNA, such that that nucleotide is switched for another nucleotide, or that nucleotide is deleted, or a single nucleotide is inserted into the DNA that causes that DNA to be different from the normal or wild type gene sequence.
Leslie G. Biesecker, M.D.”
14.8 Talking Glossary: Polymorphism (1 min)
Abstract: “Polymorphism involves one of two or more variants of a particular DNA sequence [and/or protein sequence]. The most common type of polymorphism involves variation at a single base pair [or amino acid]. Polymorphisms can also be much larger in size and involve long stretches of DNA. Called a single nucleotide polymorphism, or SNP (pronounced snip), scientists are studying how SNPs in the human genome correlate with disease, drug response, and other phenotypes.”
Transcript: “Polymorphism, by strict definitions which hardly anybody pays attention to anymore, is a place in the DNA sequence where there is variation, and the less common variant is present in at least one percent of the people of who you test. That is to distinguish, therefore, polymorphism from a rare variant that might occur in only one in 1,000 people. A polymorphism, it has to occur in at least one in 100 people. Polymorphisms could be not just single-letter changes like a C instead of T. They could also be something more elaborate, like a whole stretch of DNA, that is either present or absent. You might call that a copy number variant; those are all polymorphisms. But this is basically a general term to talk about diversity in genomes in a species.”
Francis S. Collins, M.D., Ph.D.
14.9 Talking Glossary U03: Primer (0.5 min)
National Human Genome Research Institute
Introduction: “A primer is a short, single-stranded DNA sequence used in the polymerase chain reaction (PCR) technique. In the PCR method, a pair of primers is used to hybridize with the sample DNA and define the region of the DNA that will be amplified. Primers are also referred to as oligonucleotides.”
Image: https://www.genome.gov/sites/default/files/tg/en/illustration/primer.jpg
{kind=link}
Audio: https://www.genome.gov/sites/default/files/tg/en/narration/primer.mp3
Transcript “Primer refers to a small set of nucleotides of DNA, typically 18 to 24 base pairs in length. And a primer can be used for a multitude of other experimental processes. You can use primer in PCR to target a locus to allow for amplification for further analysis. You’d use a primer for sequencing a sequencing reaction where you want to target a very specific region and then do analysis in the extension of that DNA molecule.
Stacie Loftus, Ph.D.
14.10 Talking Glossary: Promoter (1.5 min)
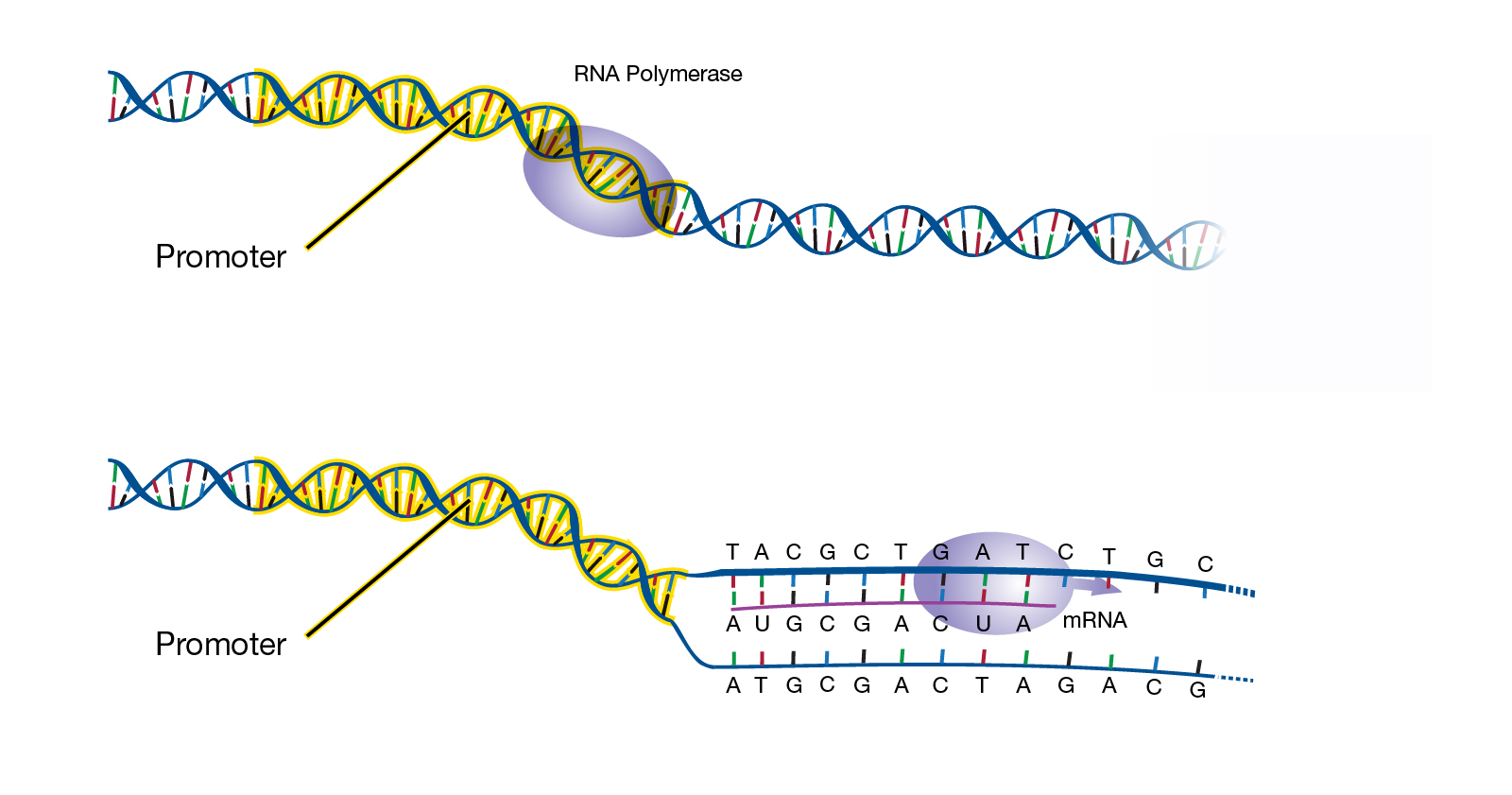
Abstract: “A promoter is a sequence of DNA needed to turn a gene on or off. The process of transcription is initiated at the promoter. Usually found near the beginning of a gene, the promoter has a binding site for the enzyme used to make a messenger RNA (mRNA) molecule.”
Audio: https://www.genome.gov/sites/default/files/tg/en/narration/promoter.mp3
Image: https://www.genome.gov/sites/default/files/tg/en/illustration/promoter.jpg
{kind=link}
Transcript: “The promoter region is the sequence typically referred to that’s right upstream or right next to where a gene is about to be transcribed. It’s the region where certain regulatory elements will bind; these are proteins that will bind to help RNA get transcribed. Now,”promoter”, the term “promoter”, can actually be a little bit of a nebulous term because it’s not very exact. There are specific sequences that are generally found within a promoter region, but sometimes people refer to even extended promoter region that might include sequences that are farther upstream of the gene that might help enhance or repress the particular gene that’s about to be transcribed in certain cell types. In general, if you think of the promoter as that piece of DNA that’s just upstream of the transcription start site of a gene, that’s pretty much what we refer to as promoters.”
Elliott Margulies, Ph.D.