Chapter 23 DNA descriptive statics - Part 1
By: Avril Coghlan
Adapted, edited and expanded: Nathan Brouwer (brouwern@gmail.com) under the Creative Commons 3.0 Attribution License (CC BY 3.0).
23.1 Preface
This is a modification of “DNA Sequence Statistics (1)” from Avril Coghlan’s A little book of R for bioinformatics.. The text and code were originally written by Dr. Coghlan and distributed under the Creative Commons 3.0 license.
23.2 Introduction
23.3 Vocabulary
- GC content
- DNA words
- scatterplots, histograms, piecharts, and boxplots
23.4 Functions
seqinr::GC()seqinr::count()
23.5 Learning objectives
By the end of this tutorial you will be able to, among other things
- Determine the GC content using GC() and obtain other summary data with count()
23.6 Preliminaries
library(compbio4all)
library(seqinr)
library(seqinr)23.7 Converting DNA from FASTA format
In a previous exercise we downloaded and examined DNA sequence in the FASTA format. The sequence we worked with is also stored as a data file within the compbio4all pa package and can be brought into memory using the data() command.
data("dengueseq_fasta")We can look at this data object with the str() command
str(dengueseq_fasta)## chr ">NC_001477.1 Dengue virus 1, complete genome\nAGTTGTTAGTCTACGTGGACCGACAAGAACAGTTTCGAATCGGAAGCTTGCTTAACGTAGTTCTA"| __truncated__This isn’t in a format we can work with directly so we’ll use the function fasta_cleaner() to set it up.
header. <- ">NC_001477.1 Dengue virus 1, complete genome"
dengueseq_vector <- compbio4all::fasta_cleaner(dengueseq_fasta)Now check it out.
str(dengueseq_vector)## chr [1:10735] "A" "G" "T" "T" "G" "T" "T" "A" "G" "T" "C" "T" "A" "C" "G" ...What we have here is each base of the sequence in a seperate slot of our vector.
The first four bases are “AGTT”
We can see the first one like this
dengueseq_vector[1]## [1] "A"The second one like this
dengueseq_vector[2]## [1] "G"The first and second like this
dengueseq_vector[1:2]## [1] "A" "G"and all four like this
dengueseq_vector[1:4]## [1] "A" "G" "T" "T"23.8 Length of a DNA sequence
Once you have retrieved a DNA sequence, we can obtain some simple statistics to describe that sequence, such as the sequence’s total length in nucleotides. In the above example, we retrieved the DEN-1 Dengue virus genome sequence, and stored it in the vector variable dengueseq_vector To obtain the length of the genome sequence, we would use the length() function, typing:
length(dengueseq_vector)## [1] 10735The length() function will give you back the length of the sequence stored in variable dengueseq_vector, in nucleotides. The length() function actually gives the number of elements (slots) in the input vector that you passed to it, which in this case in the number of elements in the vector dengueseq_vector. Since each element of the vector dengueseq_vector contains one nucleotide of the DEN-1 Dengue virus sequence, the result for the DEN-1 Dengue virus genome tells us the length of its genome sequence (ie. 10735 nucleotides long).
23.8.1 Base composition of a DNA sequence
An obvious first analysis of any DNA sequence is to count the number of occurrences of the four different nucleotides (“A”, “C”, “G”, and “T”) in the sequence. This can be done using the the table() function. For example, to find the number of As, Cs, Gs, and Ts in the DEN-1 Dengue virus sequence (which you have put into vector variable dengueseq_vector, using the commands above), you would type:
table(dengueseq_vector)## dengueseq_vector
## A C G T
## 3426 2240 2770 2299This means that the DEN-1 Dengue virus genome sequence has 3426 As occurring throughout the genome, 2240 Cs, and so forth.
23.8.2 GC Content of DNA
One of the most fundamental properties of a genome sequence is its GC content, the fraction of the sequence that consists of Gs and Cs, ie. the %(G+C).
The GC content can be calculated as the percentage of the bases in the genome that are Gs or Cs. That is, GC content = (number of Gs + number of Cs)100/(genome length). For example, if the genome is 100 bp, and 20 bases are Gs and 21 bases are Cs, then the GC content is (20 + 21)100/100 = 41%.
You can easily calculate the GC content based on the number of As, Gs, Cs, and Ts in the genome sequence. For example, for the DEN-1 Dengue virus genome sequence, we know from using the table() function above that the genome contains 3426 As, 2240 Cs, 2770 Gs and 2299 Ts. Therefore, we can calculate the GC content using the command:
(2240+2770)*100/(3426+2240+2770+2299)## [1] 46.66977Alternatively, if you are feeling lazy, you can use the GC() function in the SeqinR package, which gives the fraction of bases in the sequence that are Gs or Cs.
seqinr::GC(dengueseq_vector)## [1] 0.4666977The result above means that the fraction of bases in the DEN-1 Dengue virus genome that are Gs or Cs is 0.4666977. To convert the fraction to a percentage, we have to multiply by 100, so the GC content as a percentage is 46.66977%.
23.8.3 DNA words
As well as the frequency of each of the individual nucleotides (“A”, “G”, “T”, “C”) in a DNA sequence, it is also interesting to know the frequency of longer DNA words, also referred to as genomic words. The individual nucleotides are DNA words that are 1 nucleotide long, but we may also want to find out the frequency of DNA words that are 2 nucleotides long (ie. “AA”, “AG”, “AC”, “AT”, “CA”, “CG”, “CC”, “CT”, “GA”, “GG”, “GC”, “GT”, “TA”, “TG”, “TC”, and “TT”), 3 nucleotides long (eg. “AAA”, “AAT”, “ACG”, etc.), 4 nucleotides long, etc.
To find the number of occurrences of DNA words of a particular length, we can use the count() function from the R SeqinR package.
The count() function only works with lower-case letters, so first we have to use the tolower() function to convert our upper class genome to lower case
dengueseq_vector <-tolower(dengueseq_vector)Now we can look for words. For example, to find the number of occurrences of DNA words that are 1 nucleotide long in the sequence dengueseq_vector, we type:
seqinr::count(dengueseq_vector, 1)##
## a c g t
## 3426 2240 2770 2299As expected, this gives us the number of occurrences of the individual nucleotides. To find the number of occurrences of DNA words that are 2 nucleotides long, we type:
seqinr::count(dengueseq_vector, 2)##
## aa ac ag at ca cc cg ct ga gc gg gt ta tc tg tt
## 1108 720 890 708 901 523 261 555 976 500 787 507 440 497 832 529Note that by default the count() function includes all overlapping DNA words in a sequence. Therefore, for example, the sequence “ATG” is considered to contain two words that are two nucleotides long: “AT” and “TG”.
If you type help(‘count’), you will see that the result (output) of the function count() is a table object. This means that you can use double square brackets to extract the values of elements from the table. For example, to extract the value of the third element (the number of Gs in the DEN-1 Dengue virus sequence), you can type:
denguetable_2 <- seqinr::count(dengueseq_vector,2)
denguetable_2[[3]]## [1] 890The command above extracts the third element of the table produced by count(dengueseq_vector,1), which we have stored in the table variable denguetable.
Alternatively, you can find the value of the element of the table in column “g” by typing:
denguetable_2[["aa"]]## [1] 1108Once you have table you can make a basic plot
barplot(denguetable_2)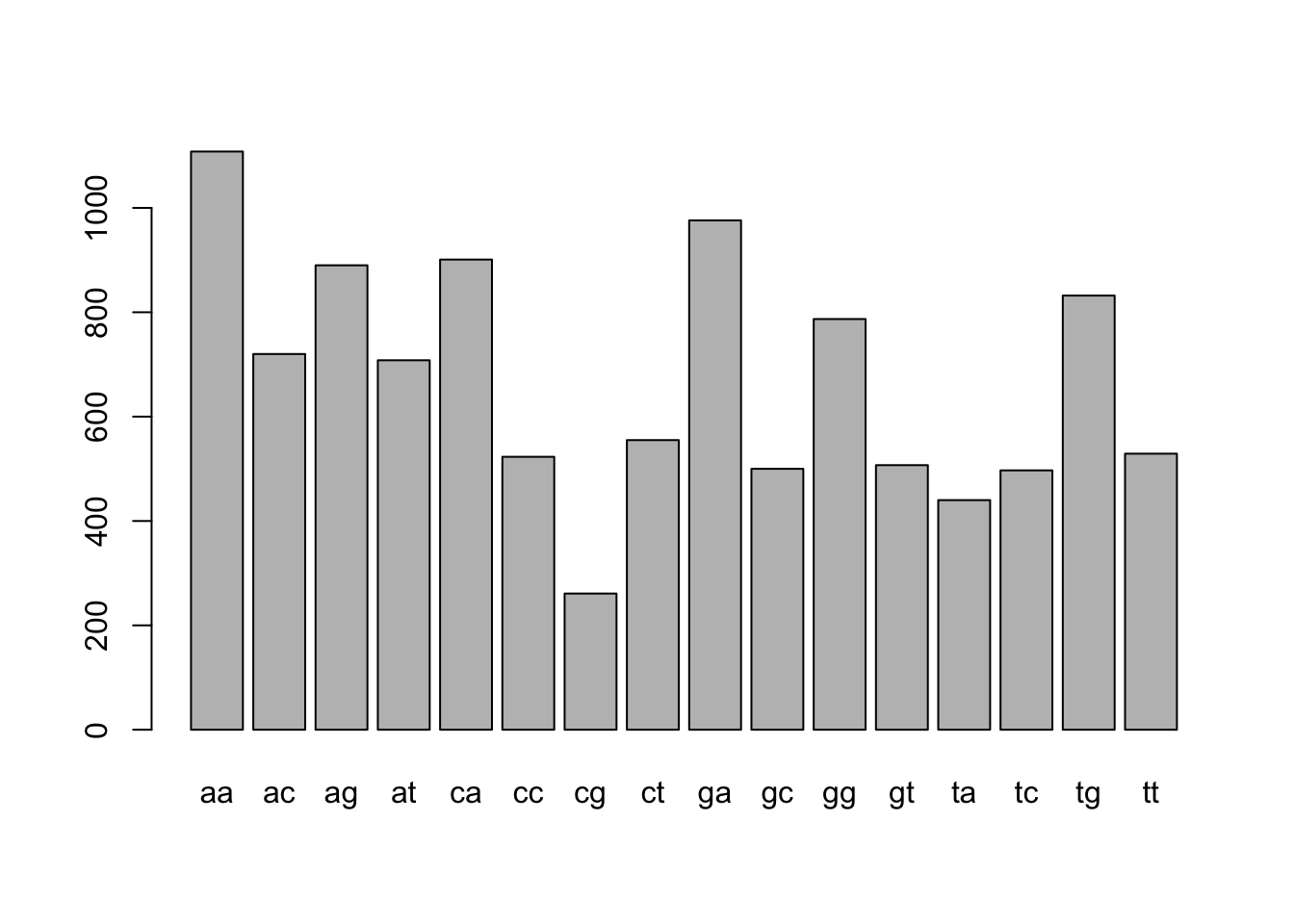
We can sort by the number of words using the sort() command
sort(denguetable_2)##
## cg ta tc gc gt cc tt ct at ac gg tg ag ca ga aa
## 261 440 497 500 507 523 529 555 708 720 787 832 890 901 976 1108Let’s save over the original object
denguetable_2 <- sort(denguetable_2)barplot(denguetable_2)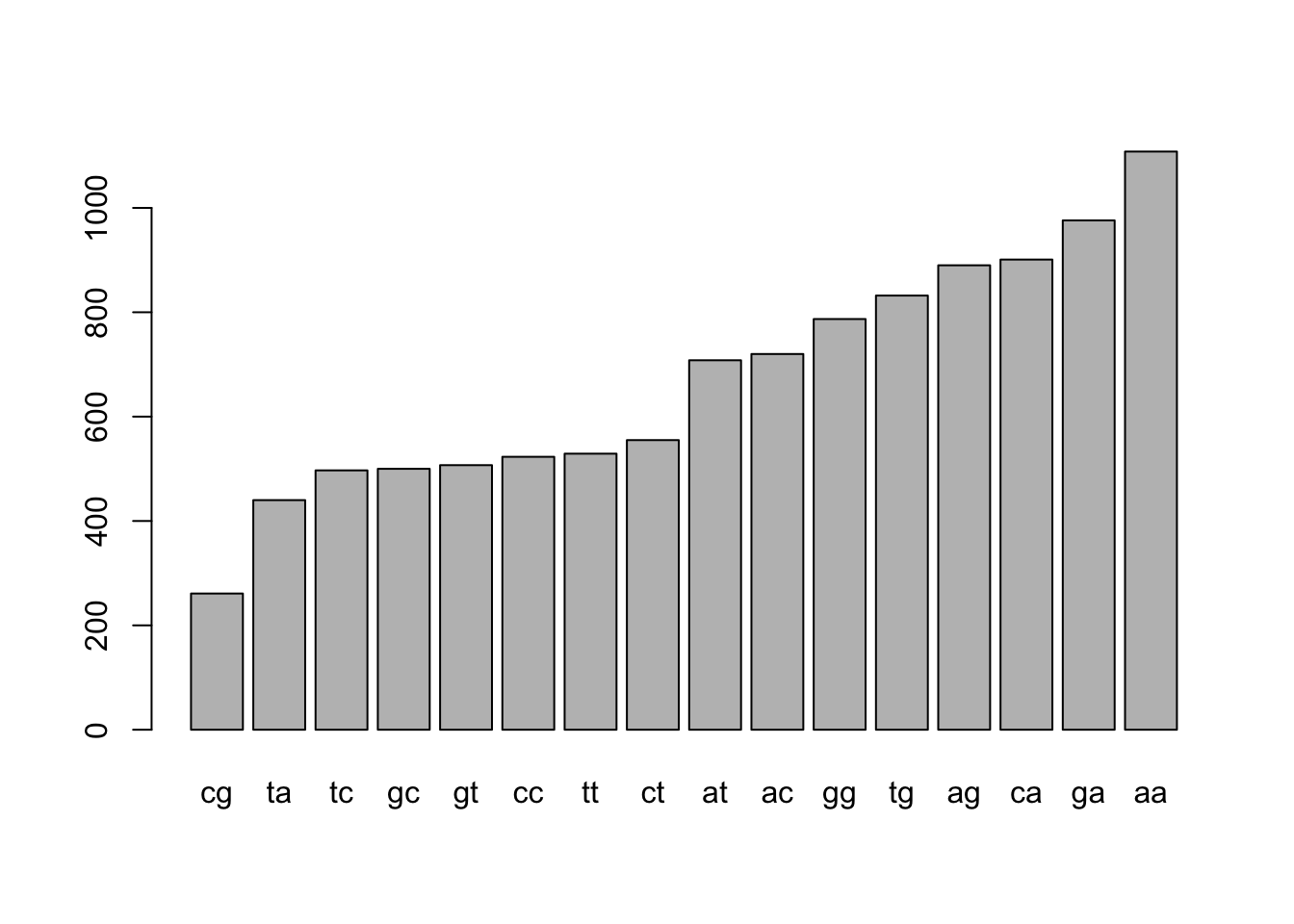
R will automatically try to optimize the appearance of the labels on the graph so you may not see all of them; no worries.
R can also make pie charts. Piecharts only really work when there are a few items being plots, like the four bases.
denguetable_1 <- seqinr::count(dengueseq_vector,1)Make a piechart with pie()
pie(denguetable_1)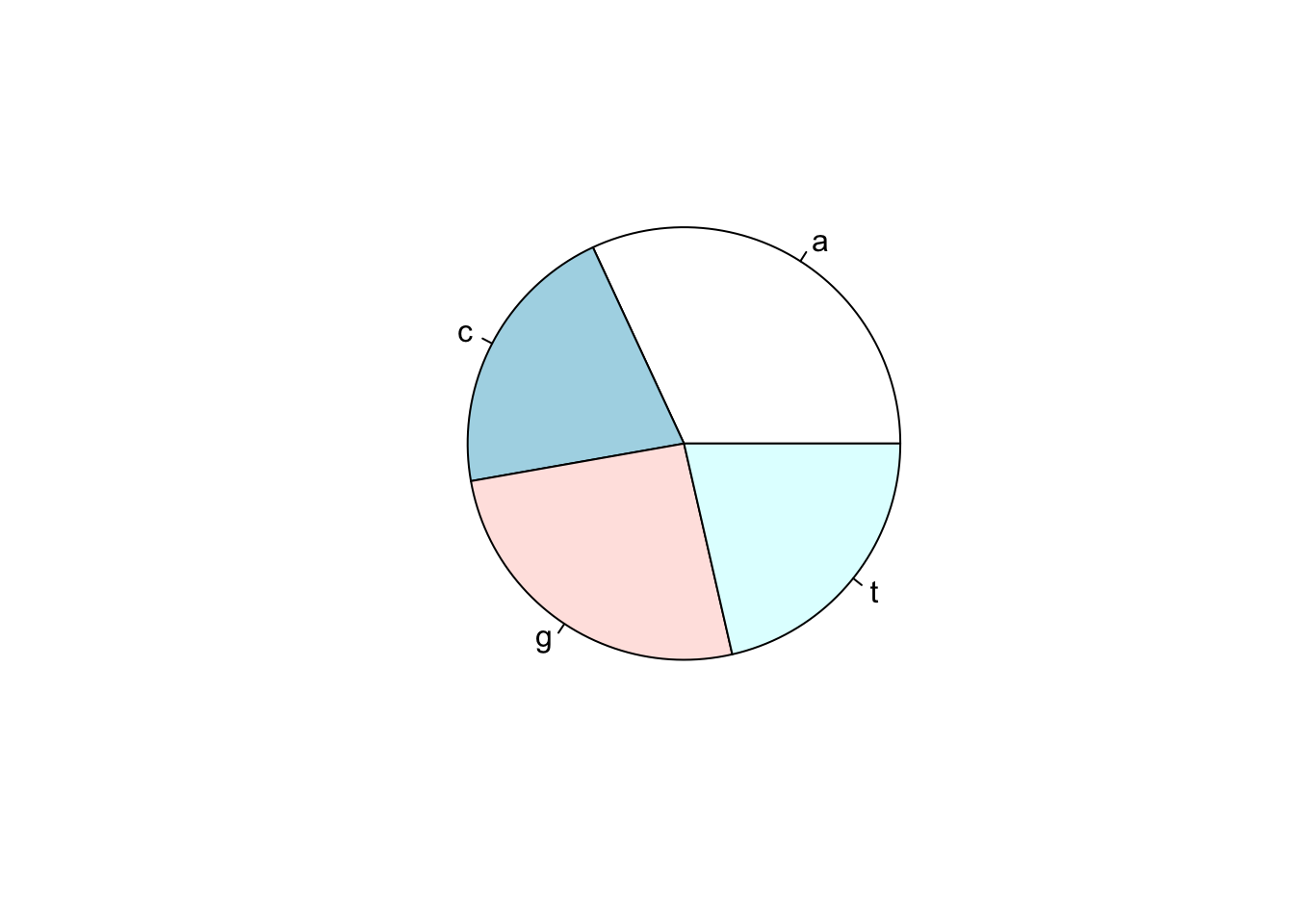
23.8.4 Summary
In this practical, have learned to use the following R functions:
length() for finding the length of a vector or list table() for printing out a table of the number of occurrences of each type of item in a vector or list. These functions belong to the standard installation of R.
You have also learnt the following R functions that belong to the SeqinR package:
GC() for calculating the GC content for a DNA sequence count() for calculating the number of occurrences of DNA words of a particular length in a DNA sequence
23.9 Acknowledgements
This is a modification of “DNA Sequence Statistics (1)” from Avril Coghlan’s A little book of R for bioinformatics.. Almost all of text and code was originally written by Dr. Coghlan and distributed under the Creative Commons 3.0 license.
In “A little book…” Coghlan noted: “Many of the ideas for the examples and exercises for this chapter were inspired by the Matlab case studies on Haemophilus influenzae (www.computational-genomics.net/case_studies/haemophilus_demo.html) and Bacteriophage lambda (http://www.computational-genomics.net/case_studies/lambdaphage_demo.html) from the website that accompanies the book Introduction to Computational Genomics: a case studies approach by Cristianini and Hahn (Cambridge University Press; www.computational-genomics.net/book/).”
23.9.1 License
The content in this book is licensed under a Creative Commons Attribution 3.0 License.
23.9.2 Exercises
Answer the following questions, using the R package. For each question, please record your answer, and what you typed into R to get this answer.
Model answers to the exercises are given in Answers to the exercises on DNA Sequence Statistics (1).
- What are the last twenty nucleotides of the Dengue virus genome sequence?
- What is the length in nucleotides of the genome sequence for the bacterium Mycobacterium leprae strain TN (accession NC_002677)? Note: Mycobacterium leprae is a bacterium that is responsible for causing leprosy, which is classified by the WHO as a neglected tropical disease. As the genome sequence is a DNA sequence, if you are retrieving its sequence via the NCBI website, you will need to look for it in the NCBI Nucleotide database.
- How many of each of the four nucleotides A, C, T and G, and any other symbols, are there in the Mycobacterium leprae TN genome sequence? Note: other symbols apart from the four nucleotides A/C/T/G may appear in a sequence. They correspond to positions in the sequence that are are not clearly one base or another and they are due, for example, to sequencing uncertainties. or example, the symbol ‘N’ means ‘aNy base’, while ‘R’ means ‘A or G’ (puRine). There is a table of symbols at www.bioinformatics.org/sms/iupac.html.
- What is the GC content of the Mycobacterium leprae TN genome sequence, when (i) all non-A/C/T/G nucleotides are included, (ii) non-A/C/T/G nucleotides are discarded? Hint: look at the help page for the GC() function to find out how it deals with non-A/C/T/G nucleotides.
- How many of each of the four nucleotides A, C, T and G are there in the complement of the Mycobacterium leprae TN genome sequence? Hint: you will first need to search for a function to calculate the complement of a sequence. Once you have found out what function to use, remember to use the help() function to find out what are the arguments (inputs) and results (outputs) of that function. How does the function deal with symbols other than the four nucleotides A, C, T and G? Are the numbers of As, Cs, Ts, and Gs in the complementary sequence what you would expect?
- How many occurrences of the DNA words CC, CG and GC occur in the Mycobacterium leprae TN genome sequence?
- How many occurrences of the DNA words CC, CG and GC occur in the (i) first 1000 and (ii) last 1000 nucleotides of the Mycobacterium leprae TN genome sequence? 1.How can you check that the subsequence that you have looked at is 1000 nucleotides long?